Der Mechanismus der Erhöhung der Inotropie (Kontraktionskraft des Herzens) durch Digoxin (Lanoxin)
Digoxin (Lanoxin) ist ein bekanntes Herzglykosid und wird seit vielen Jahren zur Behandlung verschiedener kardiovaskulärer Probleme eingesetzt, wie z. B. Herzinsuffizienz und Frequenzkontrolle bei Patienten mit Vorhofarrhythmien, die eine schnelle ventrikuläre Reaktion aufweisen.1,2 In Bezug auf die Herzinsuffizienz ist es wichtig zu beachten, dass es sich hierbei um eine systolische Herzinsuffizienz handelt und nicht um eine diastolische Herzinsuffizienz, bei der Digoxin diesen Zustand möglicherweise verschlimmern könnte.1 Diese Unterscheidung ist nicht nur klinisch wichtig, sondern bildet auch den Kontext für die vorliegende Frage.1 Als kurze Zusammenfassung: Patienten mit systolischer Herzinsuffizienz haben in der Regel ein verringertes Herzzeitvolumen (CO). Diese Verringerung des CO resultiert in erster Linie aus einer Verringerung des Schlagvolumens (SV). Das Schlagvolumen wird von einer Reihe von Faktoren beeinflusst, wie z. B. 1) der Inotropie (Kraft der Ventrikelkontraktion), 2) der Vorlast (Volumen des Blutes, das zum Herzen zurückkehrt oder die Ventrikel während der Diastole füllt) und 3) der Nachlast (Widerstand gegen den Vorwärtsblutfluss während der Systole). Daher führt alles, was einen oder mehrere dieser Faktoren erhöht, zu einer Erhöhung der SV und damit letztlich zu einer Erhöhung der CO (unter der Annahme, dass sich der Puls nicht ändert oder abnimmt).
Die meisten Kliniker erkennen an, dass die Rolle von Digoxin bei der Kontrolle von Symptomen im Zusammenhang mit systolischer Herzinsuffizienz mit seinerinotropen Wirkung oder seiner Fähigkeit, die Kontraktionskraft zu erhöhen, zu tun hat.3 Einige dieser Kliniker werden weiter sagen, dass es dies durch eine Erhöhung der intrazellulären Kalzium (Ca2+)-Konzentration tut. Das ist zwar alles richtig, aber die eigentliche Frage, die diesen Gedankengang verbindet, lautet: Wie verursacht Digoxin diesen Anstieg der zytosolischen Kalziumkonzentrationen und wie erhöht sich dadurch letztendlich die Inotropie? Um die Rolle von Digoxin bei der systolischen Herzinsuffizienz zu verstehen, muss der Kliniker die normale Physiologie des Herzzyklus verstehen, der die Kontraktion (Systole) und die Entspannung (Diastole) des Ventrikels verursacht.
Was passiert während der normalen Herzmuskelkontraktion?
Bei der ventrikulären Depolarisation (Systole) bewegt sich Natrium (Na+) in den Herzmyozyten (während der Phase 0 des kardialen Aktionspotentials; siehe Abbildung unten). Kurz darauf beginnt Kalium (K+), sich aus dem Herzmyozyten herauszubewegen, um in die extrazelluläre Umgebung zu gelangen (dies ist Phase 1 des Aktionspotentials). Während dieser Zeit steigen die zytosolischen Ca2+ -Konzentrationen bekanntermaßen durch eine Reihe von Mechanismen schnell an (dies ist Phase 2 des Aktionspotenzials).4 Wie in der zweiten Abbildung gezeigt, gelangt Ca2+ über die spannungsgesteuerten Ca2+ -Kanäle vom L-Typ in den Herzmyozyten, die die T-Tubuli des Sarkomers auskleiden (dieser Kanal wird auch als DHPR = Dihydropyridinrezeptor bezeichnet und ist der Rezeptor, an dem die Kalziumkanalblocker vom Typ „Dihydropyridin“ (d.h, Diltiazem und Verapamil) hemmen). Sobald das Ca2+ in das Zytosol gelangt, bindet es an Calmodulin, um die Ca2+/Calmodulin-abhängige Proteinkinase (auch bekannt als Myosin-Leichtkettenkinase II (MLKII oder CaMKII)) zu aktivieren.5 Sobald MLKII gebildet wurde, kann sie eine Menge Dinge tun, von denen eines die Erhöhung des zytosolischen Ca2+ ist. MLKII tut dies durch zwei Mechanismen: 1) es kann die Ryanodinrezeptoren (RyR) auf dem sarkoplasmatischen Retikulum phosphorylieren, was bewirkt, dass Ca2+ aus dem Inneren des sarkoplasmatischen Retikulums in das Zytosol (Zytoplasma) wandert, und gleichzeitig kann es 2) Phospholamban phosphorylieren, das Ca2+ während der Repolarisation über die Ca2+-ATPase (SERCA2) in das sarkoplasmatische Retikulum einlagert, um bei der nächsten kardialen Depolarisation über den RyR ausgestoßen zu werden.6,7 In den meisten Situationen arbeiten diese zusammen, jedoch kann eine sympathische Stimulation (wie bei Herzinsuffizienz) auch die Aktivität von Phospholamban während der Repolarisation erhöhen, wodurch mehr Ca2+ in das sarkoplasmatische Retikulum gelangt, das nun für die nächste Repolarisation oder das Aktionspotential bereit ist. Dies ist der Versuch des menschlichen Körpers, die Inotropie durch sympathische Stimulation zu erhöhen, insbesondere bei linksventrikulärer systolischer Herzinsuffizienz, bei der die Herzleistung beeinträchtigt ist.
Dieser Anstieg des zytosolischen Ca2+ ermöglicht dann die Bindung von Ca2+ an TroponinC, wodurch Tropomyosin bewegt wird und Myosin und Aktin miteinander interagieren können, um eine Kontraktion (oder Verkürzung des Sarkomers) zu bewirken.8,9 Je größer die Konzentration von zytosolischem Ca2+ ist, desto stärker kann dieser Prozess auftreten. Bei der Repolarisation (während der Diastole) wird nun ein Großteil des Prozesses rückgängig gemacht. Calcium wandert dann zurück in das sarkoplasmatische Retikulum oder kann über die Na+/Ca2+ -Austauschpumpe aus dem Herzmyozyten herausbewegt werden und ermöglicht so eine Entspannung der Herzmyozyten während der Diastole. Außerdem setzt dieNa+/K+-ATPase-Pumpe das Membranpotential zurück, indem sie während der Repolarisation (die Phase 3 des Aktionspotentials darstellt) 3 Na+-Ionen aus der Zelle heraus- und 2 K+-Ionen wieder in die Zelle zurückbringt.
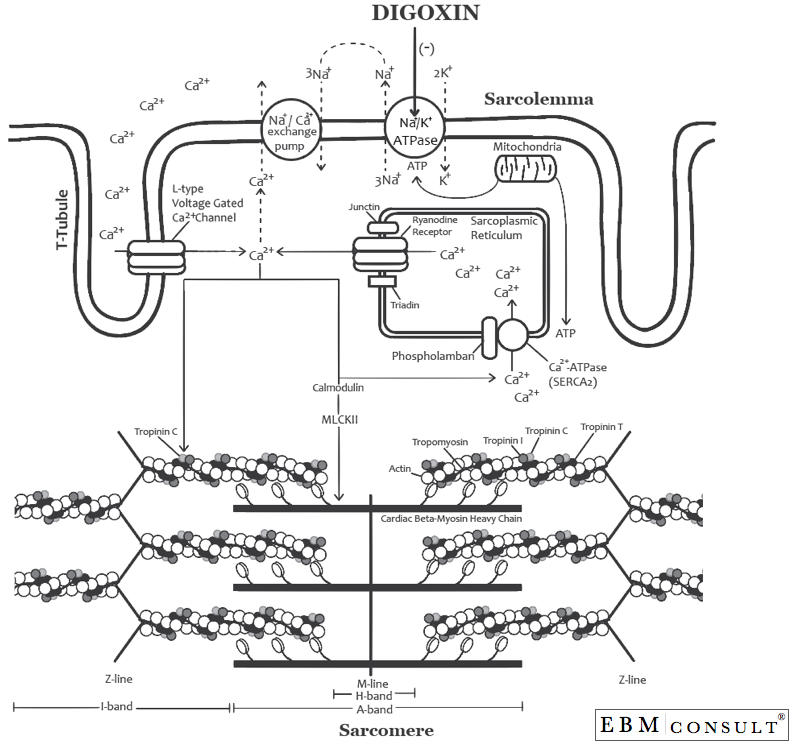
Was passiert mit diesem physiologischen Prozess, wenn dem Patienten Digoxin gegeben wird?
Wenn es im Herzen verteilt wird, bindet Digoxin an die phosphorylierte Form der Alpha-Untereinheit der Na+/K+-ATPase-Pumpe und hemmt deren Aktivität (siehe Abbildung unten).10,11 Dies führt dazu, dass die intrazelluläre orcytosolische Na+-Konzentration höher bleibt, was wiederum den Na+-Gradienten stört, der für den Betrieb der Na+/Ca2+-Austauschpumpe benötigt wird, da diese arbeitet, indem sie 3 Na+ von außerhalb des Herzmyozyten in den Myozyten bringt und im Gegenzug Ca2+ aus dem Inneren des Myozyten nehmen und nach außen transportieren würde. Daher kommt es durch Digoxin zu einer höheren Konzentration von cytosolischem Ca2+ innerhalb der Zelle, was eine stärkere Bindung an Troponin C und schließlich eine Myosin/Actin-Bindung ermöglicht, was wiederum eine größere Kontraktionskraft (oder Inotropie) zur Folge hat.
Was bedeutet das klinisch (in Bezug auf die Inotropie)?
Nun, leider nicht so viel, wie es sich anhört oder gewünscht wäre. Obwohl Digoxin einen einzigartigen und nützlichen Wirkmechanismus für Patienten mit niedrigerem CO hat, konnte die Dig-Studie keine Verringerung der Sterblichkeit bei Patienten mit Herzinsuffizienz zeigen.12 Es ist jedoch bekannt, dass Digoxin die Symptome und Krankenhausaufenthalte im Zusammenhang mit Herzinsuffizienz verringert, weshalb es bei systolischer Herzinsuffizienz im Stadium C gemäß den AHA/ACC-Richtlinien empfohlen wird.1,12 Und schließlich ist es wichtig zu bedenken, dass dieser kleine positive Effekt nur bei therapeutischen Dosierungen auftritt. Da Digoxin einen engen therapeutischen Index hat, ist eine angemessene Überwachung der Medikamentenkonzentrationen notwendig, insbesondere bei Patienten mit eingeschränkter oder veränderter Nierenfunktion und bei Beginn neuer Medikamente, die bekannte Inhibitoren der Effluxpumpe P-Glykoprotein sind.
- Jessup M, Abraham WT, Casey WT et al. 2009 focused update: ACCF/AHAGuidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American HeartAssociation Task Force on Practice Guidelines: developed incollaboration with the International Society for Heart and LungTransplantation. Circulation 2009;119:1977-2016.
- Cheng JW, Rybak I. Use of digoxin for heart failure and atrialfibrillation in elderly patients. Am J Geriatr Pharmacother 2010;8:419-27.
- Little WC, Rossi JR, Freeman GL. Comparison of effects ofdobutamine and ouabain on left ventricular contraction and relaxation in closed-chest dogs. J Clin Invest 1987:80;613-620.
- Zhang L, Kelley J, Schmeisser G et al. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteine der kardialen junktionalen sarkoplasmatischen Retikulummembran. J Biol Chem 1997;272:23389-97.
- Couchonnal LF, Anderson ME. The role of calmodulin kinase II in myocardial physiology and disease. Physiology 2008;23:151-9.
- Lanner JT, Georgiou DK, Joshi AD et al. Ryanodine receptors:structure, expression, molecular details, and function in calciumrelease. Cold Spring Harb Perspect Biol 2010;2:a003996.
- Beard NA, Wei L, Dulhunty AF. Control of muscle ryanodinereceptor calcium release channels by proteins in the sarcoplasmicreticulum lumen. Clin Exp Pharmacol Physiol 2009;36:340-5.
- Kamm KE, Stull JT. Signaling to myosin regulatory light chain in sarcomeres. J Biol Chem 2011;286:9941-7.
- Ding P, Huang J, Battiprolu PK et al. Cardiac myosin light chain kinase is necessary for myosin regulatory light chain phosphorylationand cardiac performance in vivo. J Biol Chem 2010;285:40819-29.
- Li PW, Ho CS, Swaminathan R et al. The chronic effects oflong-term digoxin administration on Na+/K(+)-ATPase activity in rattissues. Int J Cardiol 1993;40:95-100.
- Eichhorn EJ, Gheorghiade M. Digoxin. Prog Cardiovasc Dis 2002;44:251-66.
- The Digitalis Investigation Group, The effect of digoxin onmortality and morbidity in patients with heart failure, N Engl J Med 1997;336;525-533.